
Light stimulation of brain cells can recover memories in mice with Alzheimer’s disease-like memory loss, according to new research from the RIKEN-MIT Center for Neural Circuit Genetics in Japan. The rescue of memories, which changed both the structure of neurons as well as the behavior of mice, was achieved using optogenetics, a method for manipulating genetically tagged cells with precise bursts of light. This finding suggests that impaired retrieval of memories, rather than poor storage or encoding, may underlie this prominent symptom of early Alzheimer’s disease and points to the synaptic connectivity between memory cells as being crucial for retrieval.
Loss of long-term memory for specific learned experiences is a hallmark of early Alzheimer’s disease (AD) that is also exhibited by mice genetically engineered to develop AD-like symptoms. Building on their previous work that identified and activated memory cells, a group led by RIKEN Brain Science Institute and RIKEN-MIT Center Director Susumu Tonegawa has now shown that spines — small knobs on brain-cell dendrites through which synaptic connections are formed — are essential for memory retrieval in these AD mice. Moreover, fiber-optic light stimulation can re-grow lost spines and help mice remember a previous experience. The work was published in Nature on March 16.
Mouse memory is often inferred from learned behavior, in this case associating an unpleasant footshock with a particular cage. Remembering and expecting shocks causes mice to freeze in this enclosure but not in a neutral one. Compared with normal mice, AD mice exhibited amnesia and reduced freezing behavior, indicating progressive memory loss.
The engrams, or memory traces, of this particular experience are known to be located in the dentate gyrus of the hippocampus, a key brain area for memory processing. During fear conditioning, researchers used a virus to deliver a gene into the dentate gyrus, which labeled active engram cells. This allowed researchers to visually identify the neurons that made up the engram for that specific fear memory. A second virus contained a gene making only these engram neurons sensitive to light. When the engram cells were reactivated with light in the AD mice, memory of the footshock experience became retrievable and freezing behavior was restored.
Memories restored with this method faded away within a day, and the researchers next sought to understand why this happens. They noted a reduction in the number of spines as the mice aged and their Alzheimer’s disease progressed. Their waning memory for the fear training was also linked to a loss of these spines. Previous work had shown that spines grow when neurons undergo long-term potentiation, a persistent strengthening of synaptic connectivity that happens naturally in the brain but can also be artificially induced through stimulation.
Through repeated stimulation with high-frequency bursts of light to the hippocampal memory circuit in AD mice, the team was able to boost the number of spines to levels indistinguishable from those in control mice. The freezing behavior in the trained task also returned and remained for up to six days. The implication is that restoring lost spines in the hippocampal circuit facilitated retrieval of the specific fear experience and its associated freezing behavior. “We have shown for the first time that increasing synaptic connectivity within engram cell circuits can be used to treat memory loss in mouse models of early Alzheimer’s disease,” says lead author Dheeraj Roy.
Light stimulation did not boost the number of spines in normal mice or strengthen the fear memory, nor did indiscriminately shining light in the dentate gyrus result in any long-term memory improvement. Only the precise stimulation of engram cells was able to increase the number of spines and bring about the memory improvement in AD mice.
SEE ALSO: How Cholesterol-Lowering Drugs Destroy The Human Brain & Cause Dementia And Alzheimer’s Disease
“The successful retrieval of memories in AD mice by increasing the number of spines for normal memory processing only in the memory cells, rather than in a broad population of cells, highlights the importance of highly-targeted manipulation of neurons and their circuits for future therapies. This level of specificity has not yet been accomplished in current deep brain stimulation therapies,” says Tonegawa. (Reference: Dheeraj S. Roy, Autumn Arons, Teryn I. Mitchell, Michele Pignatelli, Tomás J. Ryan and Susumu Tonegawa, “Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease”, Nature, doi: 10.1038/nature17172)
.
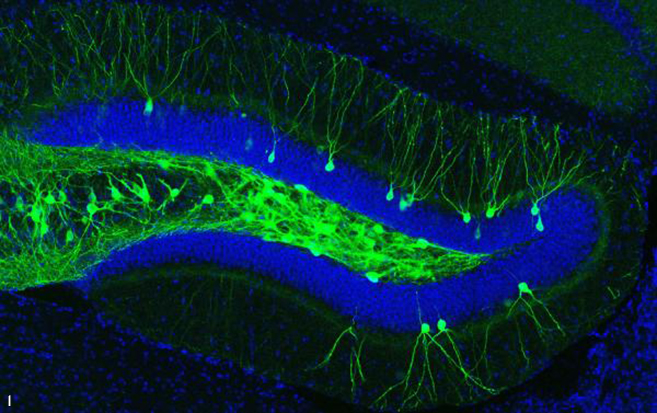
IMAGE 1 (ABOVE): ENGRAM CELLS IN THE DENTATE GYRUS OF AN A.D. MOUSE ➢ This image represents a coronal section of hippocampal dentate gyrus (DG) from a mouse model of early Alzheimer’s disease. These AD mice lack β-amyloid plaques at 7-months of age. Using these mice combined with a novel viral strategy, memory engram cells (green) for a contextual fear memory were labeled with a light-sensitive protein ChR2.
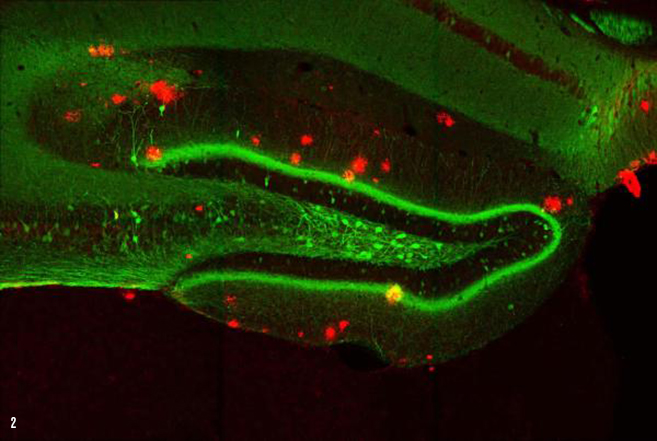
IMAGE 2 (ABOVE): ENGRAM CELLS IN 9-MONTH-OLD A.D. MOUSE WITH PLAQUES ➢ This image represents a coronal section of hippocampal dentate gyrus (DG) from a mouse model of early Alzheimer’s disease. These AD mice exhibit severe β-amyloid plaques (red) in the DG at 9-months of age. Using these mice combined with a novel viral strategy, memory engram cells (green) for a contextual fear memory were labeled.
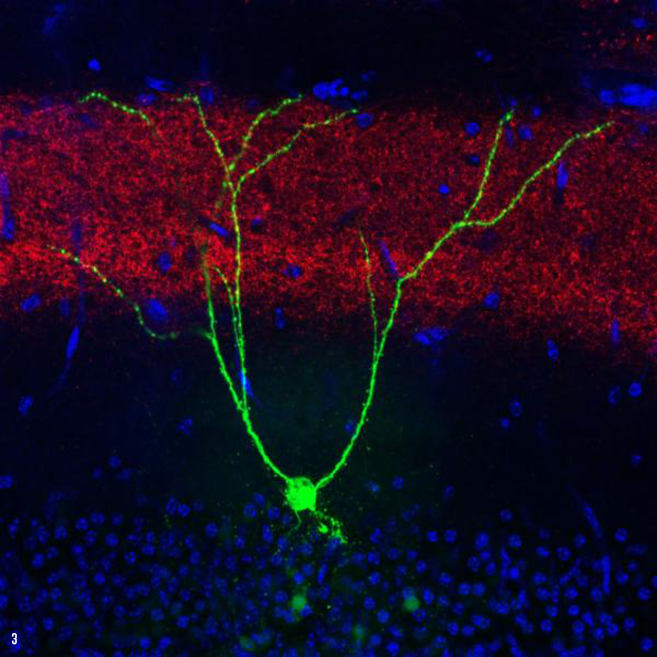
IMAGE 3 (ABOVE): ENGRAM CELL EC INPUT IN A.D. MOUSE ➢ This image depicts a single memory engram cell (green) in the hippocampal dentate gyrus (DG) region of a mouse model of early Alzheimer’s disease. To optically manipulate specific connections to these engram cells, a blue light-sensitive protein oChIEF was expressed in an upstream brain region, i.e., medial entorhinal cortical inputs (red) to the DG. The majority of DG granule cells were not active during engram labeling (blue, non-engram cells).
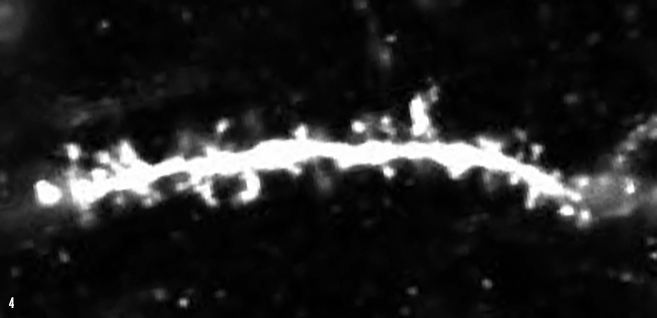
IMAGE 4 (ABOVE): DG ENGRAM CELL SPINES ➢ Synaptic input connections between upstream medial entorhinal cortex and downstream hippocampal dentate gyrus (DG) can be visualized by examining dendritic spines of DG granule cells. This image depicts dendritic spines (white protrusions) on a single DG memory engram cell in a mouse model of early Alzheimer’s disease.
And from the RIKEN labs in Japan, to the Queensland Brain Institute (QBI) at the University of Queensland in Australia, another team of researchers has made an equally remarkable breakthrough by restoring nearly 100% memory function in the brains of mice with Alzheimer’s. In the study published in the journal Science Translational Medicine, the technique uses an innovative, non-invasive, and drug-free ultrasound technology which breaks apart the neurotoxic amyloid plaques in Alzheimer’s brains which cause memory loss, dementia, and cognitive decline.
The amyloid plaques were cleared in 75% of the mice treated, and Professor Jürgen Götz, the study’s co-author, believes the new method could revolutionize Alzheimer’s treatment: “We’re extremely excited by this innovation of treating Alzheimer’s without using drug therapeutics. The ultrasound waves oscillate tremendously quickly, activating microglial cells that digest and remove the amyloid plaques that destroy brain synapses. The word ‘breakthrough’ is often mis-used, but in this case I think this really does fundamentally change our understanding of how to treat this disease, and I foresee a great future for this approach.”
SEE ALSO: The Amazing 5-Second “Peanut Butter Test” Can Detect Early Alzheimer’s With Astonishing Accuracy
“With an aging population placing an increasing burden on health systems, an important factor is cost. Other potential drug treatments using antibodies will be expensive,” adds Götz. “In contrast, this method uses relatively inexpensive ultrasound and microbubble technology, which is non-invasive and appears highly effective.”
Future Timeline writes, “The new treatment developed by QBI is able to temporarily open the blood-brain barrier — a layer that normally protects our brains from bacteria and other potential threats, but also blocks drugs from entering and therefore prohibits traditional medicine. By using ultrasound, microglial cells (which are basically a type of support cell for removing waste) were stimulated to engulf and clear toxic protein clumps, fully restoring memory functions in 75% of the mice. This was achieved without damaging brain tissue. ‘With our approach, the blood-brain barrier’s opening is only temporary for a few hours, so it quickly restores its protective role,’ Professor Götz added.” Human trials for the breakthrough treatment are set for 2017.
.
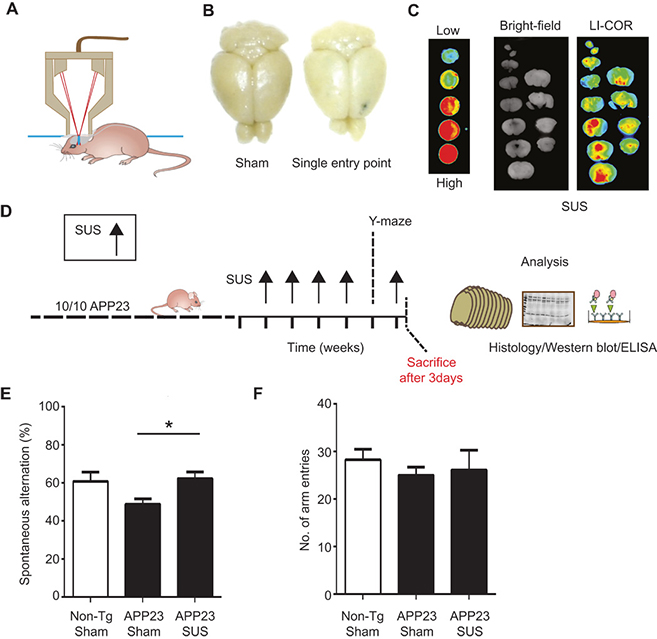
IMAGE 5 (ABOVE): ESTABLISHING SUS IN AN A.D. MOUSE MODEL ➢ (A) Setup of SUS equipment. (B and C) BBB opening by ultrasound was monitored by injecting wild-type mice with Evans blue dye that binds to albumin, a protein that is normally excluded from the brain. (B) A single entry point revealed a focal opening of the BBB in response to ultrasound treatment, with Evans blue dye able to enter the brain at this point. (C) Widespread opening of the BBB 1 hour after SUS was demonstrated with an Odyssey fluorescence LI-COR scanner of brain slices using nitrocellulose dotted with increasing concentrations of blue dye as a control. (D) Treatment scheme for the first cohort of hemizygous male Aβ plaque–forming APP23 mice (median age, 12.8 months). The mice received SUS or sham treatment for a total duration of the experiment of 6 weeks. Mice were randomly assigned to treatment groups. Using histological methods, Western blotting, enzyme-linked immunosorbent assay (ELISA), and confocal microscopy, we measured the effect of SUS treatment on amyloid pathology in mouse brain. Before the last SUS treatment, all mice were tested in the Y-maze. (E) The sequence of arm entries in the Y-maze was used to obtain a measure of alternation, reflecting spatial working memory. The percentage of alternation was calculated by the number of complete alternation sequences (that is, ABC, BCA, and CAB) divided by the number of alternation opportunities. Spontaneous alternation was restored in SUS-treated compared to sham-treated APP23 mice using non-Tg littermates as controls (n = 10 per group; one-way ANOVA followed by Dunnett’s posttest, P < 0.05). (F) Total number of arm entries did not differ between groups.
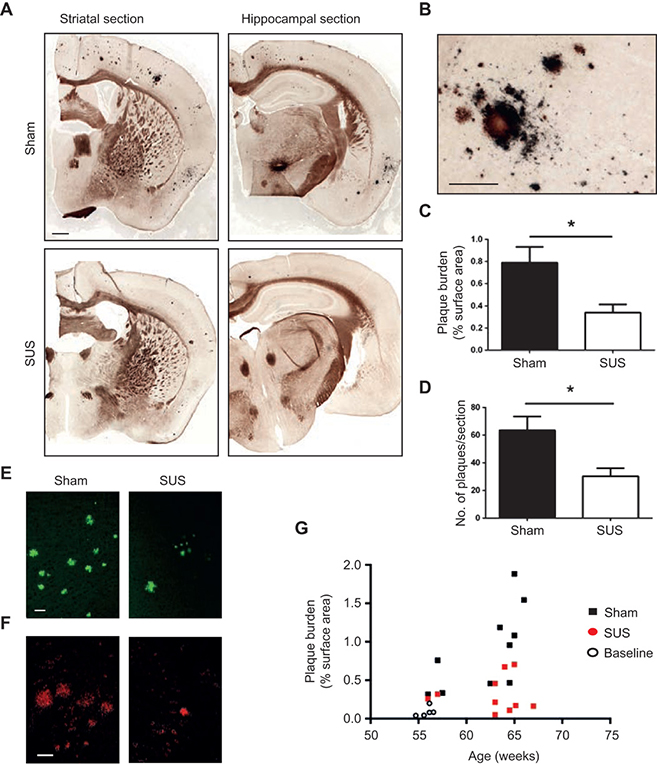
IMAGE 6 (ABOVE): SUS REDUCES SUS Aβ PLAQUES IN AN A.D. MOUSE MODEL ➢ (A and B) Representative images of free-floating coronal sections from APP23 transgenic mice (first cohort) with and without SUS treatment. Campbell-Switzer silver staining revealed compact, mature plaques (amber) and more diffuse Aβ deposits (black). A stained section at a higher magnification is shown in panel (B). (C and D) Quantification of amyloid plaques revealed a 56% reduction in the area of cortex occupied by plaques (unpaired t test, P = 0.017) and a 52% reduction in plaque number per section (t test, P = 0.014) in SUS-treated compared to sham-treated APP23 mice (n = 10 per group). (E and F) Representative sections of SUS-treated brains versus control brains stained with Thioflavin S (E) and 4G8 (F). (G) Plaque load plotted as a function of age confirmed that the SUS-treated group had significantly lower plaque load than the sham-treated group. Baseline plaque load at the onset of treatment is indicated by open circles. Scale bars, 1 mm (panel A) and 200 μm (panel B).
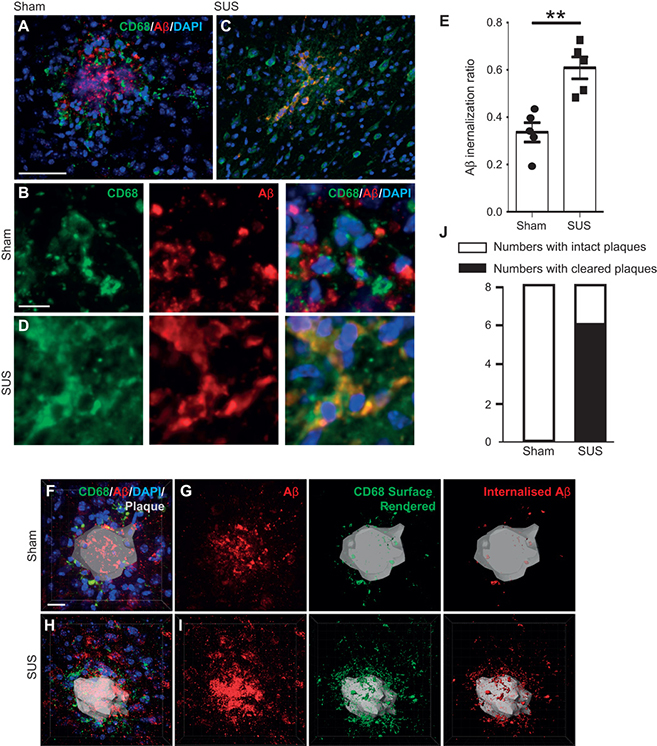
IMAGE 7 (ABOVE): MICROGLIAL PHAGOCYTOSIS AND LYSOMAL UPTAKE OF Aβ INDUCED BY SUS TREATMENT ➢ (A and B) Plaques in sham-treated animals were surrounded by lysosomal CD68-positive microglia that contained some Aβ. (C and D) In contrast, plaques in SUS-treated mouse brains were surrounded by microglia that contained significantly more Aβ in their lysosomal compartments, with some plaques appearing to be completely phagocytosed by microglia. (E) A twofold increase in microglia-internalized Aβ was observed in SUS-treated compared to sham-treated mouse brains (unpaired t test, P = 0.002). (F to I) Plaques imaged at high magnification in 3D. CD68 labeling revealed the extent of Aβ at the plaque site that was internalized by microglia into lysosomes. 4′,6-Diamidino-2-phenylindole (DAPI) was used to visualize nuclei. (J) Confocal analysis of Aβ and CD68 revealed that 6 of 8 SUS-treated mice and 0 of 8 sham-treated mice had “cleared plaques” in cortical areas, with Aβ being almost completely within microglial lysosomes (Fisher’s exact test, P = 0.007; n = 8 per group, with four sections analyzed in each case). Scale bars, 100 μm (A and C) and 10 μm (B, D, and F to I).
[Story sources: Riken, Neuroscientist News, Science Translational Medicine, Future Timeline, Science Alert. Image sources: Riken (1-4), Science Translational Medicine (5-7)]